
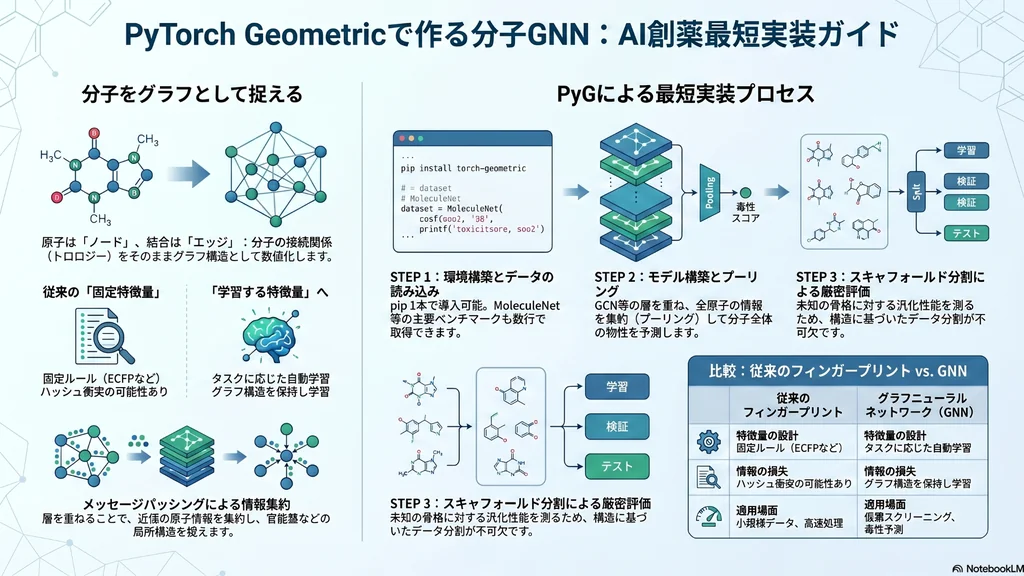
1. はじめに
新薬を一つ世に出すには、一般に10〜15年の歳月がかかると言われ、臨床試験に進んだ候補のうち約9割が承認に至らず失敗するとされています。この長く険しいプロセスを少しでも効率化しようと、近年急速に存在感を増しているのが、分子をグラフとして扱う深層学習「グラフニューラルネットワーク(GNN)」です。
本記事では、分子GNNを実装するための代表的なライブラリである「PyTorch Geometric(略称PyG)」を取り上げます。分子をなぜグラフとして表現するのかという基本概念から、インストール、データセットの読み込み、モデル実装、評価のポイントまでを、創薬研究に関心のある研究者・薬剤師の方に向けて、最短ルートで動かせるように解説します。
2. PyTorch Geometricとは:分子GNN実装の代表的なライブラリ
PyTorch Geometric(PyG)は、深層学習フレームワークPyTorchの上に構築された、グラフニューラルネットワーク専用のオープンソースライブラリです。グラフ構造を持つデータを効率的に扱うために設計されており、分子のような不規則な構造のデータと相性が良いのが特長です。GNN実装にはDGL(Deep Graph Library)やDeepChemなど他の選択肢もありますが、PyGはその中で広く使われている代表的なライブラリの一つです。
創薬の文脈でPyGが選ばれる理由は、大きく3つあります。第一に、QM9・ZINC・MoleculeNetといった創薬で定番のベンチマークデータセットを数行のコードで読み込めること。第二に、GCN・GAT・GINなど主要なGNN層が標準で実装されていること。第三に、大きさの異なる分子グラフをまとめて処理する「ミニバッチ化」やGPU高速化が組み込まれていることです。これらにより、研究者は前処理の細部に時間を取られず、モデルの設計と評価に集中できます。
なお、本記事で扱う基本的な2D分子グラフのGNNは、原子のつながり方(トポロジー)に基づく表現を学習するもので、3D配座やタンパク質との立体的な相互作用を自動的に扱うものではありません。これらを扱うには、3D座標・原子間距離・配座集合を用いる3D GNNやSE(3)/E(3)等変モデルなど、別の表現とモデル設計が必要になります。
3. なぜ分子をグラフで扱うのか:原子をノード、結合をエッジに
分子は、その構造をそのままグラフとして表現できます。具体的には、各原子を「ノード(節点)」、原子同士をつなぐ共有結合を「エッジ(辺)」として扱います。分子式は原子の種類と数しか持ちませんが、SMILES文字列は一次元表記でも結合関係・芳香族性・分岐・環などを符号化しており、分子グラフへ復元できます。グラフ表現の利点は「情報を新たに増やす」点ではなく、原子のつながり方を明示的な構造として扱い、近傍の情報を集約する計算(後述のメッセージパッシング)を自然に適用できる点にあります。
3.1. ノードとエッジが持つ特徴量
グラフの各要素には、化学的な情報を数値ベクトルとして付与します。ノード(原子)には、原子番号・電荷・価数・水素の数・芳香族性・混成軌道(sp、sp2、sp3)などを持たせます。エッジ(結合)には、結合の種類(単結合・二重結合・三重結合・芳香族結合)や、環構造に属するかどうかといった情報を持たせます。こうした特徴量の設計が、予測精度を左右する重要な要素になります。
3.2. メッセージパッシングという仕組み
GNNの中心となる計算が「メッセージパッシング」です。これは、各原子が結合した隣の原子と情報をやり取りし、自分の状態を更新していく処理を指します。1層通すと各原子は直接結合した隣接原子の情報を取り込み、層を重ねるほど、より広い範囲の原子の情報を集約していきます。たとえば3層重ねると、3結合先までの原子の情報が伝わり、官能基のような局所構造の文脈を捉えられるようになります。
最終的に、すべての原子の情報を一つにまとめる「プーリング(読み出し)」を行い、分子全体を表す一本のベクトルに変換します。このベクトルをもとに、溶解度のような連続値の予測(回帰)や、毒性の有無の判定(分類)を行うのが基本的な流れです。
4. AI創薬でGNNが注目される理由:フィンガープリントとの違い
従来の計算化学では、分子フィンガープリント(ECFPなど)や分子記述子(分子量・LogP・極性表面積など)といった、あらかじめ決められた固定の特徴量がよく使われてきました。ECFPのような円形フィンガープリントは原子・結合・芳香族性などの局所構造を反映できますが、ハッシュ化や固定長化により情報の衝突・圧縮が起こり得ること、そして特徴の設計が固定的でタスクに応じた表現学習は行わないことが特徴です。
一方GNNは、分子グラフから「そのタスクに有効な特徴量」を学習によって自動的に獲得します。両者の違いを整理すると、次のようになります。
| 観点 | 従来のフィンガープリント | GNN |
|---|---|---|
| 構造情報 | 固定ルールで局所構造を符号化。ハッシュ衝突や情報圧縮が起こり得る | グラフ上で近傍情報を集約し表現を学習 |
| 結合の扱い | 実装・設定により結合種を反映可能 | エッジ特徴量対応モデルなら結合タイプも学習に利用可能 |
| 特徴量 | 基本的に事前定義・固定 | タスクに応じた表現学習が可能 |
| 注意点 | 解釈しやすく、少量データで強い場合もある | データ量・分割・モデル設計・過学習対策に性能が依存 |
こうした性質から、GNNは仮想スクリーニング(膨大な化合物からの候補絞り込み)、合成前の毒性予測、目的の物性を持つ新規分子の生成、逆合成経路の予測など、創薬の幅広い場面で活用が進んでいます。
5. PyTorch Geometricで最短で動かす:インストールとデータ構造
5.1. インストール
基本的な利用であれば、現在は次の1行でインストールできます(公式表記はアンダースコアのtorch_geometricです)。以前はtorch-scatterなどのコンパイル済み拡張を別途入れる必要がありましたが、PyG 2.3以降はこれらが任意となり、PyTorch以外の外部ライブラリなしで最小構成から始められます。
pip install torch_geometric大規模グラフでの高速化など、追加機能が必要になった段階で、環境(CPU/CUDAのバージョン)に合わせた拡張パッケージを後から導入する流れが推奨されています。
5.2. グラフを表すDataオブジェクト
PyGでは、1つのグラフをDataというオブジェクトで表現します。最小構成は、ノード特徴量xと、どのノード同士がつながっているかを示すedge_indexの2つです。
import torch
from torch_geometric.data import Data
# どのノードとどのノードがつながっているか(COO形式)
edge_index = torch.tensor([[0, 1, 1, 2],
[1, 0, 2, 1]], dtype=torch.long)
# 各ノード(原子)の特徴量
x = torch.tensor([[-1], [0], [1]], dtype=torch.float)
data = Data(x=x, edge_index=edge_index)
print(data)
# >>> Data(x=[3, 1], edge_index=[2, 4])5.3. 創薬用データセットの読み込み
PyGは、創薬で広く使われるデータセットを標準で備えています。たとえば溶解度のデータセットESOLは、MoleculeNetから数行で読み込めます。MoleculeNetには、ESOL(溶解度)、FreeSolv(溶媒和自由エネルギー)、Tox21(毒性)、HIV(活性)などが含まれます。
from torch_geometric.datasets import MoleculeNet
# ESOL(水溶解度)データセット
dataset = MoleculeNet(root='./data', name='ESOL')
print(dataset) # 分子数・特徴量次元などを確認量子化学的な物性を扱いたい場合は、約13万分子・19種類の物性値を持つQM9データセットが定番です。こちらもfrom torch_geometric.datasets import QM9で同様に読み込めます。
自分の手元のSMILESデータから分子グラフを作りたい場合は、化学情報ライブラリのRDKitで分子を読み込み、原子・結合の特徴量を計算してDataオブジェクトに変換するのが一般的な流れです。その際は、塩の除去や構造の標準化(正規化)、重複化合物の扱いといった前処理を、学習・評価の前にそろえておくことが重要です。
6. GCNモデルの実装例:3層のグラフ畳み込み
ここでは、最も基本的なGNNであるGCN(Graph Convolutional Network)を3層重ねたモデルの例を示します。各層で原子の情報を更新し、最後に分子全体へまとめて予測する構成です。
import torch
import torch.nn.functional as F
from torch_geometric.nn import GCNConv, global_mean_pool
class GCN(torch.nn.Module):
def __init__(self, num_features, dim_h):
super().__init__()
self.conv1 = GCNConv(num_features, dim_h)
self.conv2 = GCNConv(dim_h, dim_h)
self.conv3 = GCNConv(dim_h, dim_h)
self.lin = torch.nn.Linear(dim_h, 1)
def forward(self, data):
x, edge_index, batch = data.x, data.edge_index, data.batch
# 3層のメッセージパッシング
x = self.conv1(x, edge_index).relu()
x = self.conv2(x, edge_index).relu()
x = self.conv3(x, edge_index)
# 分子全体への集約(平均プーリング)
x = global_mean_pool(x, batch)
x = F.dropout(x, p=0.5, training=self.training)
return self.lin(x)global_mean_poolが、各原子のベクトルを分子ごとに平均してまとめる役割を担います。batchは、ミニバッチ内のどの原子がどの分子に属するかを示す情報で、PyGがデータローダー側で自動的に付与してくれます。
なお、この最小例で使うGCNConvは、主にノード特徴量とグラフの接続関係(および必要に応じて1次元の重み)を利用する層であり、先に挙げた結合タイプ・芳香族結合・環所属といった多次元のエッジ特徴量(edge_attr)はそのままでは使いません。結合の種類を明示的に学習へ反映させたい場合は、NNConvのようなエッジ特徴量に対応した層やメッセージパッシング設計を用いる必要があります。
7. 学習と評価:スキャフォールド分割の重要性
学習の流れは、一般的なPyTorchと同じく「予測→誤差計算→逆伝播→更新」の繰り返しです。回帰タスクでは平均二乗誤差(MSE)などを損失関数に用います。
from torch_geometric.loader import DataLoader
train_loader = DataLoader(train_dataset, batch_size=64, shuffle=True)
model = GCN(num_features=dataset.num_features, dim_h=128)
optimizer = torch.optim.Adam(model.parameters(), lr=0.001)
criterion = torch.nn.MSELoss()
def train():
model.train()
total_loss = 0
for data in train_loader:
optimizer.zero_grad()
out = model(data)
loss = criterion(out.squeeze(), data.y)
loss.backward()
optimizer.step()
total_loss += loss.item() * data.num_graphs
return total_loss / len(train_loader.dataset)創薬で特に注意したいのが、データの分け方です。学習データとテストデータをランダムに分割すると、似た構造の分子が両方に混ざり、実際の性能より高く見積もってしまうことがあります。そこで、分子の骨格(スキャフォールド)ごとに分割するBemis-Murckoスキャフォールド分割がよく用いられます。これにより、学習時に見たことのない新規骨格に対する汎化性能を、ランダム分割よりは厳しく評価できます。
ただしスキャフォールド分割も万能ではなく、近年は実際の仮想スクリーニング条件では性能を過大評価しうるとの指摘もあります。時系列分割やクラスタ分割など複数の分け方を組み合わせて評価するのが安全です。評価指標も、ESOLのような回帰ならRMSEやMAE、Tox21・HIVのような分類ならROC-AUCやPR-AUCと、タスクに応じて選ぶ必要があります。
8. ベンチマークでの位置づけ:どの程度の精度が期待できるか
MoleculeNetなどのベンチマークでは、注意機構や結合特徴量を取り入れたGNNが、フィンガープリントベースの従来手法を多くのタスクで上回る傾向が報告されています。ただし、具体的な数値はデータ分割やモデル設定によって大きく変動するため、論文間の数字を単純比較するのは禁物です。あくまで「適切に設計すれば従来手法と同等以上が期待できる」という目安として捉え、自身の課題で再現実験を行うことが重要です。
また、吸収・分布・代謝・排泄・毒性(ADMET)の複数物性を同時に予測するマルチタスク学習も、GNNを適用できる方向の一つです。関連する物性を共有して学習することで精度が向上する場合がありますが、その効果はデータ量、物性どうしの関連性、欠測やクラス不均衡、評価分割に左右されるため、一律に有利とは限りません。
9. まとめ:分子GNNを最短で始めるために
PyTorch Geometricを使えば、分子GNNは次のステップで最短に始められます。まずpip install torch_geometricでインストールし、MoleculeNetやQM9を読み込む。次に原子をノード、結合をエッジとして分子グラフを表現し、GCNなどのGNN層でモデルを組む。そしてメッセージパッシングとプーリングで分子レベルの予測を行い、スキャフォールド分割など複数の分け方で妥当に評価する、という流れです。
GNNは、分子グラフからタスクに応じた表現を学習できるため、物性予測・毒性予測・仮想スクリーニングといった研究の支援に有用です。一方で、その予測性能はデータ品質、分割方法、適用範囲、モデル設計に依存し、予測値はあくまで候補選定の支援であって、実験的検証や専門家の判断、非臨床・臨床評価を置き換えるものではありません。PyGは、こうした分子GNNを試作・検証するための有力な実装基盤です。まずは公開データセットで小さなモデルを動かすところから、第一歩を踏み出してみてはいかがでしょうか。
参考リンク
- PyTorch Geometric 公式ドキュメント(
https://pytorch-geometric.readthedocs.io/) - PyTorch Geometric インストールガイド(
https://pytorch-geometric.readthedocs.io/en/latest/install/installation.html) - TeachOpenCADD T035:GNNによる分子物性予測チュートリアル(
https://projects.volkamerlab.org/teachopencadd/talktorials/T035_graph_neural_networks.html) - MoleculeNet:分子機械学習ベンチマーク(
https://moleculenet.org/) - Sun D. et al. “Why 90% of clinical drug development fails and how to improve it?” Acta Pharm Sin B, 2022.
関連動画(参考):YouTubeで「PyTorch Geometric molecular property prediction」と検索すると、QM9やMoleculeNetを用いた実装解説の動画を視聴できます。
免責事項
本記事は、PyTorch Geometricおよび分子GNNに関する情報提供を目的として作成されたものです。記事の内容は、公開時点で入手可能な文献・情報に基づいていますが、技術の進歩や新たな知見により、情報が変更される場合があります。記事に記載されたソフトウェアの使用結果や、それに基づく研究成果について、筆者および本ブログは一切の責任を負わないものとします。実際の創薬研究にあたっては、必ず最新の文献・公式ドキュメントを確認し、専門家の助言を得てください。
本記事は生成AIを活用して作成しています。内容については十分に精査しておりますが、誤りが含まれる可能性があります。お気づきの点がございましたら、コメントにてご指摘いただけますと幸いです。
<a href="https://amzn.to/4vjECYK" target="_blank" rel="nofollow" class="blue-link"> Amazonでこの関連書籍「グラフニューラルネットワーク ―PyTorchによる実装―」を見る</a><br>