
1. はじめに:AI創薬時代の一次スクリーニングを担うADMET-AI
近年、AI創薬の進展により、数百万から数億規模の化合物ライブラリを扱う「超大規模スクリーニング」が現実のものとなってきました。生成AIや高速ドッキングが膨大な候補化合物を生み出す一方で、それらをADMET(吸収・分布・代謝・排泄・毒性)の観点から効率よく絞り込む手法が、創薬の成否を分ける重要な鍵となっています。
そこで注目されているのが、Swansonらが2024年に発表したオープンソースのADMET予測プラットフォーム「ADMET-AI」です。本記事では、ADMET-AIの技術的基盤、最新v2の改良点、実装形態、創薬ワークフローへの組み込み方、そして導入時の留意点まで、医療関係者・研究者向けにわかりやすく解説します。
2. ADMET-AIとは?大規模ライブラリ評価に特化した機械学習プラットフォーム
ADMET-AIは、Stanford大学のKyle Swanson、James Zouらが2024年7月にBioinformatics誌で発表した、ADMET特性予測のためのオープンソース機械学習プラットフォームです。Greenstone Biosciences社が運用する公開Webサーバーと、GitHub上で配布されているPythonパッケージの両方が無償提供されています。
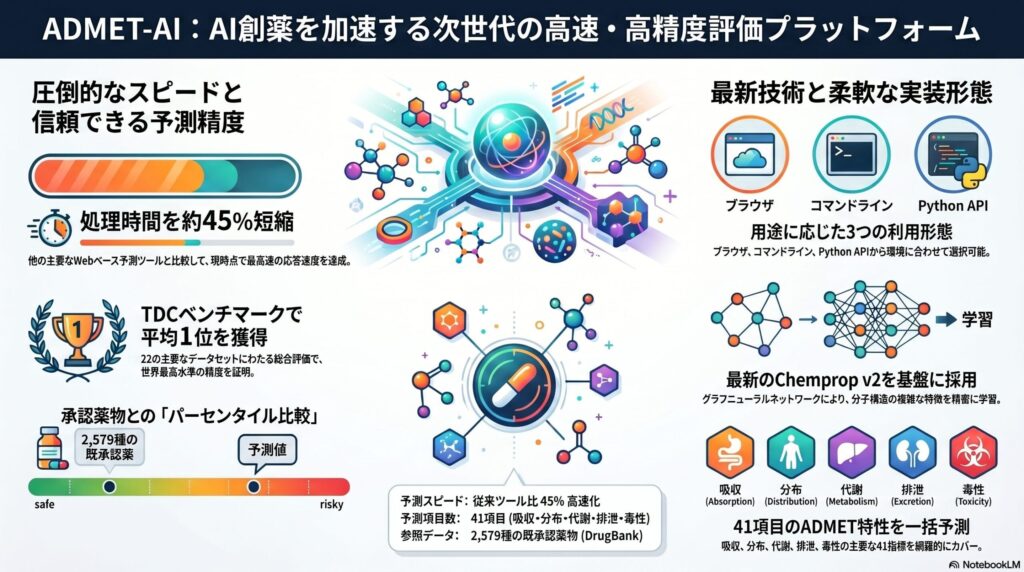
最大の特徴は「大規模化学ライブラリの評価」に特化している点です。論文によれば、ADMET-AIは公開されている他のWebベースADMET予測サーバーと比較して、約45%の処理時間短縮を実現しており、現時点で最速のWeb型ADMET予測ツールとされています。
もう一つの独自機能が「DrugBank参照セットとの比較」です。ADMET-AIは、DrugBankに登録されている2,579種類の既承認薬物に対して同じ予測を行い、ユーザーの入力分子の予測値が承認薬物群の中で何パーセンタイルに位置するかを示してくれます。これにより、抽象的な予測値が「承認薬物と比べて毒性が高い/低い」といった解釈可能な指標へと翻訳されます。
3. なぜ今ADMET-AIが選ばれるのか
創薬現場では化合物の物性を計算する手段は数多く存在しますが、その多くは「単一の特性に特化したモデル」あるいは「商用の高価なソフトウェア」でした。ADMET-AIは、無料・オープンソースでありながら最先端の精度を達成している点で、研究者にとって極めて魅力的な選択肢となっています。
特に重要なのが、Therapeutics Data Commons(TDC)のADMETベンチマークにおいて、Chempropベースのモデルが平均ランキングでトップを記録している点です。単一指標での比較ではなく、22のデータセットにわたる総合評価で最高水準を示している点が、実務での信頼性につながっています。
加えて、Webブラウザから即座に利用できる手軽さと、Python APIによる自動化の両立が可能な設計は、計算化学の専門家でなくても扱いやすく、創薬研究の入り口を広げる役割を果たしています。
4. ADMET-AIの予測性能とベンチマーク評価
ADMET-AIは、以下の観点から定量的に評価されています。
- TDC ADMETリーダーボードで平均ランキング1位(22データセット総合)
- Webサーバーの応答速度で、次点ツール比45%の高速化を達成
- 1,000分子までの一括予測がWebインターフェース上で可能
- DrugBank 2,579薬物との百分位(パーセンタイル)比較を自動表示
- ATCコード(解剖治療化学分類)による参照セット絞り込みに対応
これらの数値は、論文(Bioinformatics 2024)で公式に報告されているものです。「速度」と「精度」のバランスが取れている点が、大規模スクリーニングへの活用を後押ししています。
ベンチマークの内訳としては、吸収(Caco-2透過性、ヒト腸管吸収、Pgp阻害、バイオアベイラビリティ、Lipophilicity、水溶解度)、分布(血液脳関門透過、血漿タンパク結合率、分布容積)、代謝(CYP2C9・2D6・3A4の阻害および基質)、排泄(半減期、肝クリアランス、ミクロソーム代謝クリアランス)、毒性(LD50、hERG阻害、Ames変異原性、薬物誘発性肝障害)といった、創薬に不可欠な項目が網羅されています。
5. 技術的基盤:Chempropグラフニューラルネットワーク
ADMET-AIの予測エンジンを支えているのが、Chempropと呼ばれるグラフニューラルネットワーク(GNN)です。Chempropは、分子の構造をグラフとして扱い、原子間のメッセージパッシングを通じて分子全体の特徴を学習する手法を実装したオープンソースパッケージです。
5.1. 指向性メッセージパッシング(D-MPNN)の仕組み
Chempropの核となる技術は「指向性メッセージパッシングニューラルネットワーク(D-MPNN)」です。具体的には、SMILES文字列をRDKitで分子グラフに変換し、各原子に初期埋め込みを与えた後、結合方向に沿ってメッセージを伝播させていきます。最終的に原子埋め込みを集約して分子全体の埋め込みを生成し、順伝播型ニューラルネットワーク(FFN)で目的のADMET特性へ変換します。
この方式により、結合の方向性や局所的な化学環境を捉えた高精度な予測が可能になります。ADMET-AI v1では、Chempropの出力に加えてRDKitが計算する200の物理化学特徴量を結合する「Chemprop-RDKit」構成を採用していました。
5.2. ADMET-AI v2のアップデート:高速化と互換性向上
2026年現在、最新版のADMET-AI v2はChemprop v2を採用しており、RDKitフィンガープリントを内部で使わない構成へと移行しています。Chemprop v2自体が実行時間で約2倍の高速化、メモリ使用量で約3分の1への削減を達成しているため、ADMET-AI v2もより快適に動作します。複数GPUへのスケーリングや最新PyTorchとの互換性も改善されました。
v2では各分子に対して、RDKitによる8つの基本物性値と41のADMET特性予測がセットで出力されます。学習データはTDCから収集された41のADMETデータセットで、TDCベンチマークグループの22項目を中核としています。なお、モデルが再学習されているため、v1とv2では予測値が完全には一致しない点に注意が必要です。
6. ADMET-AIの使い方:3つの利用形態
ADMET-AIは、用途に応じて3つの利用形態が用意されています。公式リポジトリはhttps://github.com/swansonk14/admet_ai で公開されています。
第一に、Webインターフェース(admet.ai.greenstonebio.com)です。SMILES文字列の直接入力、CSVファイルのアップロード、インタラクティブな分子描画ツールが利用でき、最大1,000分子までを一括処理できます。出力にはサマリープロット、レーダーチャート、詳細予測テーブルが含まれ、DrugBank参照セットはATCコードで絞り込み可能です。
第二に、コマンドラインツールです。admet_predict --data_path input.csv --save_path output.csv のような形でCSVを一括処理でき、数万〜数百万分子規模のバッチ予測に向いています。サーバーで一晩動かすような大規模ジョブにも適しています。
第三に、Python APIです。from admet_ai import ADMETModel でモデルをロードし、SMILESリストを渡すだけで予測結果が得られます。RDKitやPyTorchベースの他の創薬パイプラインへの統合が容易で、自社の自動化ワークフローに組み込む際に重宝します。
7. 他のADMET予測ツールとの比較と使い分け
ADMET-AIをより活用するため、代表的な他ツールとの違いを整理します。
- ADMETlab 3.0:119の予測項目を備えるWeb型プラットフォーム。項目数の多さで網羅性に強み
- SwissADME:物理化学特性とDrug-likeness評価に特化。古典的ルール(Lipinski等)に基づく解釈が中心
- Simulations Plus ADMET Predictor:175以上の特性を予測する商用プラットフォーム。CYP代謝部位や動力学パラメータ予測まで含む産業用ツール
- ADMET-AI:41項目を高精度・高速に予測し、DrugBankとの百分位比較で実務的解釈をサポート
実務での使い分けとしては、まずADMET-AIで大規模ライブラリ全体を一次スクリーニングし、上位候補に絞ってからADMETlab 3.0や商用ツールで詳細評価する、といった多段階運用が効率的です。ADMET-AIはAPI連携が容易なため、生成AIによる分子設計パイプラインの直後に組み込む使い方も推奨されます。
8. 実務導入における課題と今後の展望
ADMET-AIは強力なツールですが、導入時にはいくつかの留意点があります。最大の課題は「汎化性」で、学習データに含まれない新規化学空間(例:マクロ環状化合物、PROTACなど)では予測信頼性が低下する可能性があります。出力された予測値は絶対的な真実ではなく、あくまで意思決定を支援する一指標として扱うことが重要です。
また、深層学習モデル特有の「ブラックボックス性」も課題です。なぜその予測値になったかを機構論的に説明することが難しいため、合成判断や安全性評価では、必ず実験的検証と専門家のレビューを組み合わせる必要があります。
今後の展望としては、分子構造に加えて細胞画像や配列情報を統合する「マルチモーダル学習」、化学・生物学を横断する「基盤モデル(Foundation Models)」、複数組織でのプライバシー保護学習を可能にする「連合学習(Federated Learning)」といった方向性が、ADMET予測の次のフロンティアとして注目されています。ADMET-AIは、これらの新技術を取り込みながら進化を続けていくと予想されます。
9. まとめ:ADMET-AIで創薬スクリーニングを加速する
ADMET-AIは、Chempropグラフニューラルネットワークを基盤とし、TDCの41 ADMETデータセットで学習されたオープンソースの予測プラットフォームです。Webインターフェース、CLI、Python APIの3形態で提供され、DrugBank 2,579薬物との百分位比較によって、予測値を実務的な意思決定に翻訳できる点が他にない強みとなっています。
大規模化合物ライブラリの一次スクリーニングを高速・高精度に行えるADMET-AIは、AI創薬時代の標準ツールの一つとして定着しつつあります。本記事を参考に、ぜひ自身の研究テーマでもWebインターフェースから試してみてください。生成AIが生み出す膨大な候補化合物を、ADMET-AIで賢く絞り込むことで、創薬プロジェクトの効率を一段引き上げることができるはずです。
参考文献・リンク
- ADMET-AI論文(Bioinformatics 2024):https://academic.oup.com/bioinformatics/article/40/7/btae416/7698030
- ADMET-AI公式Webサーバー:https://admet.ai.greenstonebio.com/
- ADMET-AI GitHubリポジトリ:https://github.com/swansonk14/admet_ai
- Chemprop GitHubリポジトリ:https://github.com/chemprop/chemprop
- Chemprop v2論文(J. Chem. Inf. Model.):https://pubs.acs.org/doi/10.1021/acs.jcim.5c02332
- TDC ADMETベンチマークグループ:https://tdcommons.ai/benchmark/admet_group/overview/
免責事項
本記事は、ADMET-AIに関する情報提供を目的として作成されたものです。記事の内容は、公開時点で入手可能な文献・情報に基づいていますが、技術の進歩や新たな知見により、情報が変更される場合があります。記事に記載されたソフトウェアの使用結果や、それに基づく研究成果について、筆者および本ブログは一切の責任を負わないものとします。実際の創薬研究や臨床応用にあたっては、必ず最新の文献・公式ドキュメントを確認し、専門家の助言を得てください。
本記事は生成AIを活用して作成しています。内容については十分に精査しておりますが、誤りが含まれる可能性があります。お気づきの点がございましたら、コメントにてご指摘いただけますと幸いです。
Amazonでこの関連書籍「AI創薬:医薬品研究開発におけるディープラーニング・機械学習の応用」を見る