
1.はじめに
現代の創薬プロセスにおいて、コンピューターを用いたシミュレーションは欠かせない存在となっています。その中でも、薬剤(配位子)が標的タンパク質(受容体)にどれだけ強く結びつくかを数値化する「結合自由エネルギー計算」は、新薬候補の選別において極めて重要な指標です。
本記事では、世界的に利用されている分子動力学シミュレーションソフト「GROMACS」で、この計算を劇的に効率化するツール「gmx_MMPBSA」について解説します。医療従事者や創薬研究者の方々に向けて、その仕組みから活用法までをステップ・バイ・ステップで紐解いていきましょう。
2. 創薬における「結合自由エネルギー」の重要性とgmx_MMPBSAの役割
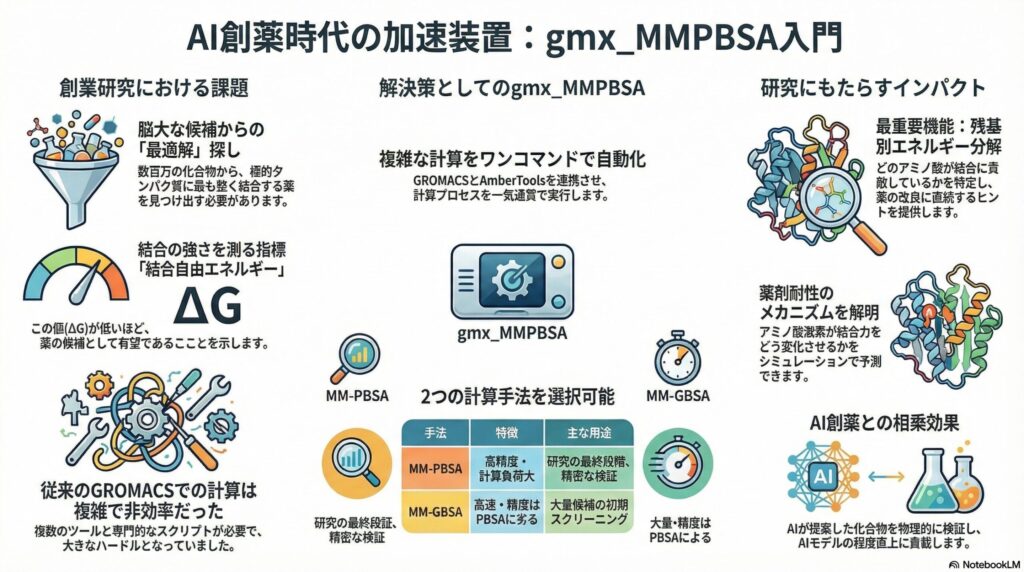
創薬の現場では、数万から数百万もの化合物の中から、病気の原因となるタンパク質にぴたっとはまる「鍵」を探し出す作業が行われます。この「はまり具合」の強さを科学的に示したものが「結合自由エネルギー(ΔG)」です。この値がマイナスに大きければ大きいほど、その薬は標的と強く結合し、高い薬効が期待できることを意味します。
これまで、GROMACSという優れたシミュレーションソフトを使用している研究者にとって、この自由エネルギーを簡便に計算することは一つの高いハードルでした。なぜなら、計算手法自体が非常に複雑で、複数のツールを組み合わせる必要があったからです。そこに登場したのが、今回ご紹介する「gmx_MMPBSA」です。
gmx_MMPBSAは、分子の動きを追跡したデータ(軌跡)から、物理化学的な計算を一気通貫で行ってくれる強力なツールです。これにより、これまで専門的なスクリプトを組まなければ得られなかった高度な解析結果を、標準的な手順で、かつ再現性高く取得することが可能になりました。まさに創薬研究の「加速装置」と言えるでしょう。
3. 計算手法の基礎:MM-PBSAとMM-GBSAの違いを理解する
gmx_MMPBSAがサポートしている主な計算手法には、「MM-PBSA」と「MM-GBSA」の2種類があります。これらは「エンドステート法」と呼ばれ、シミュレーションの最初と最後(結合状態)の状態を見ることで、計算時間を抑えつつ実用的な精度を得る手法です。
まず「MM-PBSA」ですが、これは「ポアソン・ボルツマン方程式」という物理学の理論を用いて、水の中での分子の振る舞いを精密に計算します。精度が高い反面、計算にはそれなりのコンピューターパワー(CPU負荷)が必要です。より確かな結論が欲しい、研究の最終段階などで重宝される手法です。
一方の「MM-GBSA」は、計算をより簡略化した「一般化ボルン法」を用います。精度はPBSAに一歩譲るものの、計算速度が圧倒的に速いのが特徴です。膨大な数の新薬候補をざっとランク付けしたい場合や、スクリーニングの初期段階で非常に有効な手段となります。
4. 解析を始めるための準備:必要な環境とファイルのセットアップ
gmx_MMPBSAを使用するためには、いくつかの「道具」を揃える必要があります。まず、核となるのは「GROMACS」と、アメリカの著名な研究グループが開発した「AmberTools」です。gmx_MMPBSAは、いわばこの2つの巨人の肩の上に立って動くツールと言えます。
インストールは、Pythonというプログラミング言語のパッケージ管理システム「pip」を使って行います。医療統計などでPythonに触れている方なら、pip install gmx-MMPBSA という短いコマンド一つで導入できる手軽さに驚かれるかもしれません。Windows環境でも、仮想環境(venv)を作ることで安定して動作させることが可能です。
解析に際して準備するファイルは4つです。分子の設計図である「トポロジーファイル(.top)」、分子の形を示す「構造ファイル(.gro)」、分子の動きを記録した「軌跡ファイル(.xtc)」、そして解析対象(タンパク質と薬など)を指定する「インデックスファイル(.ndx)」です。これらが揃えば、準備は完了です。
5. 実践ステップ:gmx_MMPBSAを実行し結果を得る手順
準備が整ったら、いよいよ計算の実行です。gmx_MMPBSAの素晴らしい点は、複雑な計算プロセスを一つのコマンドラインで完結できる点にあります。ユーザーは「設定ファイル(.inファイル)」に、どの手法(PBSAかGBSAか)を使うか、塩濃度(生理的食塩水に近い150mMなど)はどうするかを記述します。
実行コマンドを入力すると、ツールが自動的にAmberToolsを呼び出し、GROMACSのデータを処理し始めます。この過程で、静電気的な引き合い(静電相互作用)や、分子同士の物理的な接触(ファンデルワールス力)、さらに水に溶けやすさ(溶媒和エネルギー)といった各要素が個別に計算されていきます。
計算が終わると、FINAL_RESULTS_MMPBSA.dat というファイルが生成されます。ここに、最終的な結合自由エネルギーの値がまとめられています。単に「結合するかどうか」だけでなく、「どのエネルギー成分が結合に大きく寄与しているか」まで詳細に可視化されるため、薬の構造をどう改良すべきかのヒントが得られます。
6. 結果の解釈と創薬への応用:残基別分解能の魅力
gmx_MMPBSAの機能の中で、特に医療・創薬研究において価値が高いのが「残基別エネルギー分解(Decomposition)」です。これは、タンパク質を構成するアミノ酸一つひとつが、薬とどれくらい強く相互作用しているかを個別に算出する機能です。
例えば、ある薬の結合力が弱かったとします。この機能を使えば、「35番目のバリンというアミノ酸とはうまく噛み合っているが、120番目のグルタミン酸とは反発している」といった具体的な情報が手に入ります。これは、有機合成化学者が「薬のこの部分に枝を付ければ、120番目の反発を解消できるはずだ」という戦略を立てる際の強力な根拠になります。
また、変異体タンパク質に対する耐性の解析にも役立ちます。特定の薬が効かなくなった耐性ウイルスやがん細胞において、どのアミノ酸の変異が結合エネルギーを低下させているかをシミュレーションで特定できれば、耐性を克服する次世代薬の開発スピードを飛躍的に高めることができるのです。
7. 解析の精度を高めるためのベストプラクティスと注意点
シミュレーションは万能ではありません。gmx_MMPBSAで信頼性の高いデータを得るためには、いくつかの注意点があります。まず最も重要なのは、入力となるシミュレーション(MD)自体が「平衡状態」に達しているかを確認することです。分子が激しく動いている不安定な状態のデータを使っても、正しい答えは得られません。
次に、「サンプリング」の長さです。短時間のシミュレーションデータだけでは、たまたまその瞬間の状態を切り取っただけになる可能性があります。統計的に意味のある平均値を得るためには、十分な長さの軌跡データを使用することが推奨されます。
また、pH(水素イオン指数)の設定にも注意が必要です。体内の環境、例えば胃の中と血液中ではpHが異なります。これによってタンパク質の電気的な状態(プロトネーション状態)が変わるため、目的とする組織の環境に合わせて、適切にシミュレーションの条件を設定することが、実臨床に即した結果を得る鍵となります。
8. まとめと今後の展望:AI創薬とのシナジー効果
gmx_MMPBSAは、GROMACSユーザーにとって「自由エネルギー計算」というブラックボックスを解き放つ、非常に優れたツールです。その高い利便性と詳細な解析機能は、創薬研究の効率を劇的に向上させ、より安全で効果的な薬剤の創出に寄与することでしょう。
近年注目されているAI(人工知能)を用いた創薬においても、このツールの役割は重要です。AIが提案した膨大な数の化合物候補を、gmx_MMPBSAによって物理学的な観点から検証し、質の高い学習データをフィードバックすることで、AIの予測精度をさらに高めるという相乗効果が期待されています。
医療の進歩は、こうした地道な計算技術の積み重ねによって支えられています。gmx_MMPBSAというツールを使いこなし、分子レベルでの深い洞察を得ることは、次世代の医療を切り拓く大きな一歩となるはずです。本記事が、皆様の研究や業務の一助となれば幸いです。
免責事項
本記事に掲載された情報は、執筆時点での学術的知見およびソフトウェアの仕様に基づいています。シミュレーション結果の妥当性や特定環境での動作を保証するものではありません。本ツールの利用により生じたいかなる損害についても、当方では責任を負わないものとします。実際の研究や開発への適用に際しては、公式ドキュメントを参照し、専門家による適切な検証を行ってください。
本記事は生成AI (Gemini) を活用して作成しています。内容については十分に精査しておりますが、誤りが含まれる可能性があります。お気づきの点がございましたら、コメントにてご指摘いただけますと幸いです。
Amazonでこの関連書籍「インシリコ創薬: 計算創薬の基礎から実例まで」を見る