
1. はじめに:なぜ今、医療研究にMDシミュレーションが必要なのか
現代の創薬研究において、コンピュータ上で分子の動きを再現する「分子動力学(MD:Molecular Dynamics)シミュレーション」は欠かせない技術となりました。従来の実験手法だけでは捉えきれない、タンパク質と薬物候補分子の結合プロセスを微視的に観察できるからです。
しかし、これまでのMDは膨大な計算時間を必要とし、専門的なスーパーコンピュータ環境が必須でした。この高いハードルを打ち破り、デスクトップPCやクラウド環境で高速・手軽にシミュレーションを可能にしたのが「OpenMM」です。本記事では、AI創薬の強力な武器となるOpenMMについて、ステップバイステップで解説します。
2. ステップ1:OpenMMとは何か?―軽量で高性能な計算エンジンの正体
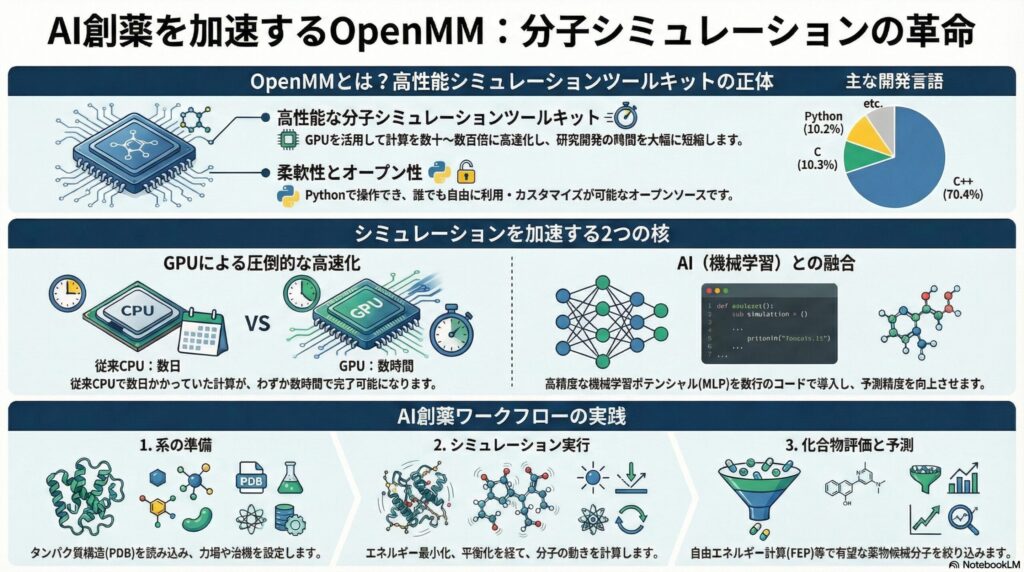
OpenMMは、一言で言えば「分子の動きを計算するための高性能な道具箱」です。オープンソースとして公開されており、誰でも自由に利用・カスタマイズが可能です。最大の特徴は、計算を高速化するための「GPU(画像処理装置)」というパーツを最大限に活用するように設計されている点にあります。
一般的に、分子のシミュレーションには数百万回、数千万回の計算反復が必要です。OpenMMは、この計算をGPUに最適化させることで、従来のCPU(中央演算処理装置)を用いた計算に比べて数十倍から数百倍の速度向上を実現しました。これにより、数日かかっていた計算が数時間で完了するようになったのです。
また、OpenMMは「Python(パイソン)」という、AI開発で最も普及しているプログラミング言語で操作できます。これにより、最新のAIモデルとシミュレーションをシームレスに組み合わせることが可能となり、創薬研究の自動化や高度化を強力に後押ししています。
3. ステップ2:GPU加速がもたらす医療研究へのインパクト
OpenMMの真骨頂は、その圧倒的なスループット(処理能力)にあります。特に、NVIDIA社が提供する「MPS(Multi-Process Service)」という技術を活用することで、1枚のGPUで複数のシミュレーションを同時に効率よく走らせることができます。これは、限られた研究リソースを最大化するために極めて有効です。
具体的な例を挙げると、中規模なタンパク質系(約2万原子)のシミュレーションにおいて、最新の最適化手法を適用することで、単位時間あたりの計算量を2倍以上に引き上げることが可能です。これは、より多くの化合物候補を、より短期間で評価できることを意味します。
医療現場に近い研究者にとって、この「速度」は「精度の向上」にも直結します。短時間で多くのシミュレーションを行えば、統計的な信頼性が高まり、より確実性の高い薬物候補の絞り込みが可能になるからです。OpenMMは、計算の「量」を「質」へと変換する装置と言えるでしょう。
4. ステップ3:AI(機械学習)との融合が拓く新しいシミュレーション
近年のOpenMMにおける最大のトピックは、バージョン8から強化された「機械学習ポテンシャル(MLP)」との統合です。従来のMDでは「力場」と呼ばれる簡略化された数式を用いて分子間の力を計算してきましたが、これには精度に限界がありました。
一方、量子化学計算という手法を用いれば非常に高精度な計算が可能ですが、計算負荷が重すぎて、タンパク質のような巨大分子には適用できませんでした。このジレンマを解決するのがAIです。AIに量子化学計算の結果を学習させることで、高精度かつ高速な計算を実現したのがMLPです。
OpenMM-MLという拡張機能を使えば、ANI-2xやMACEといった最新のAIモデルを数行のコードでシミュレーションに組み込めます。これにより、これまで見逃されていた微細な分子間相互作用を正確に記述できるようになり、より「効く」薬のデザインが可能になります。
5. ステップ4:実践!創薬ワークフローでの具体的な活用事例
では、実際にOpenMMはどのように創薬の現場で使われているのでしょうか。代表的なのは「自由エネルギー摂動(FEP)」計算です。これは、ある化合物から別の化合物へ変化させた際の結合力の差を精密に予測する手法です。
OpenMMは、このFEP計算のエンジンとして非常に優秀です。複数の計算条件(ウィンドウ)を並列で実行することで、化合物のランキングを迅速に作成できます。これにより、合成実験を行う前に「どの分子が最も有望か」を高精度に予測し、無駄な実験コストと時間を大幅に削減できます。
また、タンパク質の構造が柔軟に変化する様子をシミュレーションする「構造基盤ドラッグデザイン(SBDD)」にも活用されます。静的な結晶構造だけでは見えない「隠れたポケット」をOpenMMで発見し、そこに合致する新しいタイプの阻害剤を設計する研究が盛んに行われています。
6. ステップ5:導入の第一歩―環境構築とプロトタイピングの進め方
OpenMMを導入するのは、意外なほど簡単です。「Conda(コンダ)」と呼ばれるパッケージ管理システムを使えば、コマンド数回で最新の環境を構築できます。専門のIT部門に依頼しなくても、研究者自身の手元のワークステーションで開始できるのが大きな魅力です。
まずは、PDB(Protein Data Bank)から取得したタンパク質構造ファイルを読み込み、標準的な力場を適用して数ナノ秒のシミュレーションを回してみることから始めましょう。OpenMMの公式ドキュメントには豊富なサンプルコードが用意されており、Pythonの基礎知識があればスムーズに習得できます。
注意点として、OpenMMの性能をフルに発揮させるには、比較的新しい世代のNVIDIA製GPU(Voltaアーキテクチャ以降)を推奨します。また、大規模な系を扱う場合は、16GB以上のビデオメモリを持つGPUを選択することで、エラーを回避し安定した運用が可能になります。
7. おわりに:AI駆動型創薬の基盤としてのOpenMM
OpenMMは単なる計算ソフトの枠を超え、AIと物理シミュレーションを融合させる「プラットフォーム」へと進化しました。医療・創薬研究において、これまで「勘」や「経験」に頼っていた部分を、客観的なデータと高度な予測によって補完する時代が来ています。
今後、量子コンピューティングや分散型学習との連携も期待されており、OpenMMの重要性はさらに高まっていくでしょう。最新のテクノロジーを柔軟に取り入れ、創薬プロセスのデジタルトランスフォーメーション(DX)を推進するために、OpenMMは最適な選択肢の一つです。
免責事項
本記事の内容は、公開時点での技術情報を基に作成されています。OpenMMの利用や実装にあたっては、公式ドキュメントを参照し、各自の責任において実行してください。本記事の情報を用いたことにより生じたいかなる損害についても、筆者および当ラボは一切の責任を負わないものとします。
本記事は生成AI (Gemini) を活用して作成しています。内容については十分に精査しておりますが、誤りが含まれる可能性があります。お気づきの点がございましたら、コメントにてご指摘いただけますと幸いです。
Amazonでこの関連書籍「分子動力学シミュレーションの基礎理論」を見る