
1.はじめに:なぜ医療・創薬に「前処理」が重要なのか
現代の創薬プロセスにおいて、コンピューター上でタンパク質の動きを再現する「分子動力学(MD)シミュレーション」は欠かせないツールとなっています。新薬候補化合物が標的タンパク質にどのように結合し、その構造をどう変化させるかを可視化できるからです。
しかし、シミュレーションの結果が信頼できるかどうかは、計算を開始する前の「準備(前処理)」で決まると言っても過言ではありません。生体内の環境をいかに正確にコンピューター内に再現できるか、その具体的な手法をステップ・バイ・ステップで解説します。
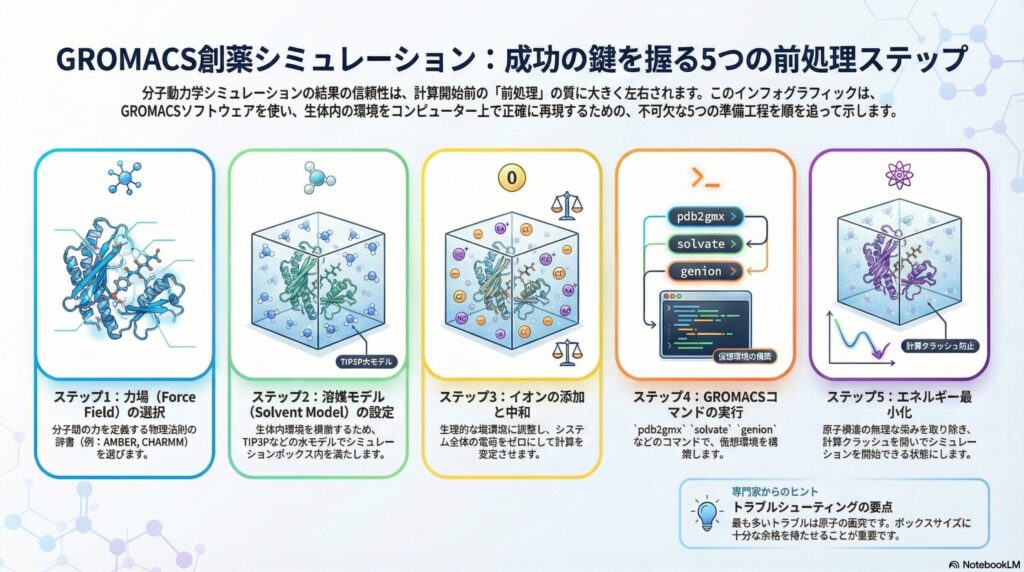
2.ステップ1:力場(Force Field)の選択—分子の「性格」を決めるルール
シミュレーションを始める際、最初に行うのが「力場」の選択です。力場とは、原子と原子の間にどのような力が働くかを規定した数理モデルのセットです。いわば、コンピューター内の仮想世界における「物理法則の辞書」のようなものです。
医療研究でよく使われる力場には、タンパク質や核酸の記述に優れた「AMBER(アンバー)」や、細胞膜などの脂質層を含む解析に強い「CHARMM(チャーム)」があります。研究対象が血中の酵素なのか、細胞膜上の受容体なのかによって、最適な辞書を使い分けることが精度向上の第一歩となります。
3.ステップ2:溶媒モデルの設定—生体内の「水」を再現する
タンパク質は、真空中で機能しているわけではありません。常に水分子に囲まれ、その影響を受けて形を保っています。そのため、シミュレーションボックス内を水分子で満たす「溶媒化」が必要です。ここで使われるのが「水モデル」と呼ばれるテンプレートです。
最も一般的なのは「TIP3P」というモデルで、計算スピードと精度のバランスが非常に優れています。より精密な温度変化や密度を議論したい場合には、少し計算は重くなりますが「TIP4P」といったモデルが選ばれることもあります。これらを選択することで、試験管内(in vitro)に近い環境を再現できます。
4.ステップ3:イオンの添加と中和—生理的条件を整える
生体内の環境は、純粋な水ではなく、ナトリウム($Na^+$)や塩素($Cl^-$)などのイオンが溶け込んだ「生理食塩水」のような状態です。また、タンパク質自体も電荷(プラスやマイナスの電気)を帯びており、システム全体が電気的に不安定な状態にあることが少なくありません。
そこで、特定のイオンを添加してシステム全体の電荷をゼロ(中和)にし、さらに生理的な塩濃度(約150 mM)に調整します。これにより、静電的な相互作用が安定し、実際の体内での挙動により近いシミュレーションが可能になります。このステップを怠ると、計算が途中で破綻する原因となります。
5.ステップ4:GROMACSによる実践的なコマンド操作
準備が整ったら、実際のソフトウェア「GROMACS」でコマンドを入力していきます。まず、pdb2gmxというコマンドでPDB(タンパク質構造データ)をGROMACS形式に変換します。この際、先ほど選んだ力場と水モデルを指定します。
次に、editconfでシミュレーションを行う箱(ボックス)の大きさを決め、solvateで水を満たします。最後にgenionを使って、計算に必要なイオンを配置します。これらの操作は一見複雑ですが、一つひとつの工程が「体内の環境を精密に模倣する」という目的を持っています。
6.ステップ5:エネルギー最小化—構造の「歪み」を取り除く
全ての配置が終わった直後の構造には、原子同士が異常に接近しているなどの「無理な力(歪み)」がかかっていることがよくあります。そのままシミュレーションを開始すると、計算が爆発(クラッシュ)してしまうため、「エネルギー最小化」という作業を行います。
これは、システム全体のエネルギーが最も低く、安定した状態を探すプロセスです。例えるなら、くしゃくしゃになった衣服をアイロンできれいに整えるような作業です。この工程を経て初めて、タンパク質が自然に動き出すための「スタートライン」に立つことができるのです。
7.専門家によるトラブルシューティングとアドバイス
前処理において最も多いトラブルは、水分子やイオンがタンパク質と重なってしまう「クラッシュ」です。これを防ぐには、ボックスサイズに十分な余裕を持たせることが重要です。また、特殊な修飾(リン酸化など)を受けたタンパク質を扱う場合は、標準的な力場では対応できないケースもあります。
その際は、追加のパラメータ設定が必要になります。計算結果が理論値と大きく異なる場合は、前処理の段階で力場が正しく適用されているか、トポロジーファイル(設計図)を入念にチェックしてください。地道な確認こそが、高品質なシミュレーションデータの源泉となります。
8.まとめ:正確な前処理が信頼性の高い創薬研究を支える
GROMACSを用いたMDシミュレーションは、適切な力場、溶媒、イオンの設定という「前処理」があって初めて意味を成します。これらは単なる計算の準備ではなく、生物学的な妥当性をコンピューター上に構築する極めて重要なプロセスです。
正しい手順で作成されたシミュレーションモデルは、疾患のメカニズム解明や新薬の分子設計において、強力なエビデンスを提供してくれるはずです。本記事が、医療現場や研究室でシミュレーションを活用する際の一助となれば幸いです。
免責事項
本記事の情報は、執筆時点での一般的な手法に基づくものです。シミュレーションの結果や特定の研究への適用について、筆者および当ラボは一切の責任を負わないものとします。実際の研究に際しては、最新の公式ドキュメントを確認し、ご自身の責任において実施してください。
本記事は生成AI (Gemini) を活用して作成しています。内容については十分に精査しておりますが、誤りが含まれる可能性があります。お気づきの点がございましたら、コメントにてご指摘いただけますと幸いです。
Amazonでこの関連書籍「できるGoogle NotebookLM 可能性は無限大!自分専用AIノート活用法 (できるシリーズ)」を見る